
Генетика — это наука о наследственности и изменчивости организма. Раздел генетики, который занимается изучением наследственности и изменчивости человека с точки зрения патологии, называется медицинской генетикой. Основными задачами медицинской генетики являются:
- Изучение наследственных форм патологии, их этиологии, патогенеза, совершенствование диагностики, разработка методов профилактики и лечения.
- Изучение причин и механизмов наследственно детерминированной предрасположенности и резистентности к различным (в том числе и инфекционной природы) заболеваниям.
- Изучение роли и значения генетического аппарата в развитии реакций адаптации, компенсации и явлениях декомпенсации.
- Подробное всестороннее изучение процессов мутагенеза и антимутагенеза, их роли в развитии болезней.
- Изучение ряда общебиологических проблем: молекулярногенетических механизмов канцерогенеза, роли генетического аппарата в явлениях тканевой несовместимости, аутоиммунных реакциях организма и др.
Понятия «наследственные болезни» и «врожденные заболевания» далеко не однозначны. Врожденными называют любые заболевания, проявляющиеся сразу после рождения ребенка. Они могут быть наследственными и ненаследственными. К числу наследственных болез-ней относятся лишь те, в основе которых лежат структурные изменения в генетическом материале. Одни из них клинически проявляются уже в первые дни после рождения, другие —в юношеском, зрелом, а иногда и в пожилом возрасте. Ненаследственные болезни обуслов-лены действием неблагоприятных факторов среды на развивающийся плод в период беременности и не затрагивают его генетический аппарат.
В медицинской генетике выделяют еще одно понятие — фенокопии. Фенокопия представляет собой клинический синдром, возникающий под влиянием факторов внешней среды в период эмбрионального развития, сходный по своим проявлениям с наследственным заболева-нием, но имеющий негенетическую природу возникновения. Например, такие аномалии, как «волчья пасть», «заячья губа», могут быть и наследственно обусловленными (синдром Патау), и ненаследственными, возникающими в результате нарушения эмбрионального разви-тия. Гипотиреоз наследуется как аутосомно-рецессивный признак, но может встречаться и как фенокопия у людей, проживающих в рай-онах, где питьевая вода бедна йодом. Ранняя глухота может наследоваться как рецессивный или доминантный признак, а может встре-чаться как фенокопия у детей, рожденных женщинами, переболевшими во время беременности краснухой. Таким образом, фенокопии представляют собой заболевания, внешне похожие на наследственные болезни, но не связанные с изменением генотипа. Причинами фенокопии могут быть: – кислородное голодание плода (внутриутробная гипоксия), вызывающее развитие серьезных дефектов структуры мозга и черепа, микроцефалию; – эндокринные нарушения в организме беременной женщины (вероятность рождения больного ребенка у такой женщины примерно в 2,5 раза выше); – инфекционные заболевания беременной женщины (токсоплазмоз, краснуха, сифилис и др.), особенно в ранний период беременности, вызывающие в значительном проценте случаев (до 60–70 %) тяжелые уродства (микроце-фалию, глухонемоту, расщелину мягкого неба и др.); – тяжелая психическая травма и длительные эмоциональные перенапряжения жен-щины в период беременности; – лекарственные препараты, обладающие цитотоксическим или антиметаболическим действием; – хронический алкоголизм родителей (пороки развития у детей непьющих родителей составляют около 2 %, у умеренно пьющих — до 9 %, у сильно пьющих — 74 %) и др.
Оглавление
Классификация наследственных болезней.
Наследственные болезни – это патологические состояния, в основе которых лежит изменение наследственного материала.
| ХРОМОСОМНЫЕ | МОНОГЕННЫЕ | МУЛЬТИФАКТОРИАЛЬНЫЕ (ПОЛИГЕННЫЕ) |
| А. Аномалии количества половых хромосом: Синдром Шерешевского-Тернера, Синдром Клайнфельтера, Синдром трисомии Х и др. | А. Аутосомно-доминантные: Синдром Марфана, Ахондроплазия, Анемия Минковского — Шоффара, Полидактилия и др. | А. ЦНС: Эпилепсия, Шизофрения, и др. |
| Б. Аномалии количества аутосом: Болезнь Дауна, Синдром Эдвардса, Синдром Патау и др. | Б. Аутосомно — рецессивные: Фенилкетонурия, Галактоземия, Целиакия, Муковисцидоз, Адреногенитальный синдром и др. | Б. Сердечно-сосудистые: Ревматизм, Атеросклероз и др. |
| В. Структурные аномалии хромосом: Синдром «кошачьего крика» и др. | В. Х-сцепление рецессивные: Гемофилия, Миопатия Дюшенна, Ихтиоз и др. | В. Кожные: Атонический дерматит, Псориаз, и др. Аллергический альвеолит и др. |
| Г. Х-сцепленные доминантные: Витамин Д- резистентный рахит, Коричневая окраска эмали зубов и др. | Г.Выделительной системы: Нефриты, Мочекаменная болезнь, Энурез, и др. | |
| Д.Пищеварительной системы: Язвенная болезнь, Цирроз печени, Неспецифический язвенный колит и др. |
Причиной развития моногенных заболеваний является поражение генетического материала на уровне молекулы ДНК, в результате чего повреждается только один ген. Наследуются они в соответствии с законами Менделя. И по типу наследования могут быть разделены на:
- аутосомно-доминантные,
- аутосомно-рецессивные,
- сцепленные с Х -хромосомой.
- сцепленные с Y-хромосомой.
Причиной развития хромосомных заболеваний являются аномалии в количестве или структуре хромосом, то есть мутации. Заболева-ния, которые вызываются геномными и хромосомными мутациями, называются хромосомными болезнями. Патологические изменения возникают как при потере генетического материала, так и при добавлении новых хромосом.
Болезни с наследственной предрасположенностью (мультифакториальные) являются полигенными и для их проявления требуется влия-ние определенных факторов внешней среды. Признаками этих заболеваний являются:
- высокая чистота среди населения.
- выраженный клинический полиморфизм.
- сходство клинических проявлений у пробанда и родственников.
- возрастные отличия.
- половые отличия.
- различная терапевтическая эффективность.
- несоответствие закономерностей наследования менделевским моделям.
Моногенные заболевания.
Общая характеристика моногенные заболевания.
Причинами многих наследственных моногенных заболеваний у человека являются генные мутации. Первая группа заболеваний связана с качественными изменениями белковых молекул, т.е. наличием у больных аномальных белков (например, аномальные гемоглобины), что обусловлено мутациями структурных генов. Другая группа заболеваний характеризуется количественными изменениями содержания нормального белка в клетке (повышение или снижение), что обусловлено, чаще всего, мутациями функциональных генов, т.е. связано с нарушениями регуляции работы генов. Эти нарушения могут осуществляться на различных уровнях:
- претранскрипционном (осуществляется путем увеличения или уменьшения числа копий гена),
- траскрипционном (генетические дефекты в спейсерах, интронах, транспозонах, регуляторных белках могут приводить к нарушению транскрипции всего гена, обусловливающие изменение объема синтеза соответствующего белка),
- процессинга и сплайсинга про-и-РНК (нарушения на уровне разрушения неинформативных участков про-и-РНК и сплавления информативных участков),
- трансляционном (нарушения на уровне непосредственной сборки белковой молекулы в рибосоме)
- посттрансляционном (нарушения на уровне образования вторичной, третичной и четвертичной структуры белковой молекулы).
Фенотипически генные мутации проявляются как наследственные болезни обмена веществ — ферментопатии. Вещества, накапливаю-щиеся в результате отсутствия или снижения активности ферментов, либо сами оказывают токсическое действие, либо включаются в цепи вторичных обменных процессов, в результате которых образуются токсические продукты. В настоящее время описано около 3 тыс. наследственных болезней обмена веществ. Общая частота генных болезней в популяциях людей составляет 2—4%.
Генные болезни классифицируют по их фенотипическому проявлению: болезни, связанные с нарушением аминокислотного, углеводного, липидного, минерального обменов и обмена нуклеиновых кислот, нарушения свертывания крови, гемоглобинопатии и др.
Характеристика моногенных заболеваний, связанных с нарушением аминокислотного обмена.
Наиболее часто встречающимися болезнями, связанными с нарушением аминокислотного обмена являются: фенилкетонурия и альбинизм. В норме аминокислота фенилаланин (ФА) с помощью фермента фенилаланингидроксилазы превращается в аминокислоту — тирозин, который в свою очередь под действием фермента тирозиназы может превращаться в меланин (пигмент). При нарушении активности этих ферментов развиваются два наследственных заболевания человека: фенилкетонурия и альбинизм.

- Фенилкетонурия (ФКУ) встречается в человеческих популяциях с относительно высокой частотой — 1:10000. Заболевание наследуется по аутосомно-рецессивному типу — больные являются рецессивными гомозиготами (аа). Ген, мутации которого приводят к заболеванию (ответственный за синтез фермента фенилаланингидроксилазы) картирован (12q22-q24), идентифицирован и секвенирован (определена последовательность нуклеотидов).Фенилаланин принадлежит к числу незаменимых аминокислот, Только часть ФА используется для син-теза белков; основное количество этой аминокислоты окисляется до тирозина. Если неактивен фермент фенил-аланингидроксилаза, то ФА не превращается в тирозин, а накапливается в сыворотке крови в больших количествах и превращается в фенилпировиноградную кисло-ту (ФПВК), выделяющуюся с мочой и потом, вследствие чего от больных исходит «мышиный» запах. Дети с фенилкетонурией рождаются здоровыми, но в первые же недели жизни у них появляются клинические признаки заболевания. ФПВК является нейротропным ядом, в результате чего развивается повышенная возбудимость, гиперрефлексия и повышенный тонус мышц, тремор, судорожные эпилептифор-мные припадки. Позже присоединяются нарушения высшей нервной деятельности, умственная отсталость, микроцефалия. У больных наблюдается слабая пигментация вследствие нарушения синтеза меланина.
Диагностика заболевания осуществляется биохимическими методами: еще до развития клинической картины наблюдается выделение ФПВК с мочой, а в крови определяется высокое содержание фенилаланина. Довольно эффективным методом лечения ФКУ является диетотерапия — снижение содержания в пище ребенка аминокислоты фенилаланина. Для предотвращения необратимых поражений мозга лечение необходимо начинать с первых недель жизни, проводить до 7-10 лет и постоянно следить за содержанием фенилаланина в крови. Мозг взрослого человека устойчив к высоким концентрациям ФПВК.
- Альбинизм встречается в разных популяциях с разной частотой — от 1:5000 до 1:25000. Он наследуется по аутосомно-рецессивному типу. В основе этого заболевания лежит мутация гена, при которой нарушается активность фермента тирозиназы, превращающего аминокислоту тирозин в пигмент меланин. Основными клиническими проявлениями альбинизма являются отсутствие меланина в клетках кожи (молочно-белый ее цвет), волосах (очень светлые волосы), радужной оболочке глаза и повышенная чувствительность к УФ-облучению, которое вызывает воспалительные заболевания кожи. У больных на коже отсутствуют какие-либо пигментные пятна, снижена острота зрения. Фенотипические признаки выражены уже у новорожденного. Диагностика заболевания не представ-ляет затруднений. Лечение не разработано.
- Алкаптонурия встречается довольно редко (3—5:1000000). Наследуется по аутосомно-рецессивному типу. Алкаптонурия является следствием генетического дефекта оксидазы, катализирующей превращение гомогентизиновой кислоты в малеилацетоуксусную. Ген картирован — 3q2. В основе этой патологии лежат отложения гомогентизиновой кислоты в соединительной ткани, создающие пигментацию цвета охры. Большое количество кислоты выводится с мочой, что приводит к ее потемнению при стоянии на воздухе. Клинические проявления заболевания начинаются в возрасте 40 лет и старше и характеризуются поражением суставов конечностей и позвоночника.
Диагностика заболевания не представляет затруднений (клиника и биохимические методы).
Характеристика моногенных заболеваний, связанных с нарушением углеводного обмена.
Наиболее частыми наследственными дефектами нарушения обмена углеводов являются галактоземия и мукополисахаридозы.
- Галактоземия встречается с частотой примерно 1:100000 новорожденных. Наследуется по аутосомно-рецессивному типу. В основе этого заболевания лежит недостаточность фермента галактозо-1-фосфатуридилтрансферазы, переводящей галактозо-1-фосфат в уридин-дифосфогалактозу. Галактоза поступает в организм с пищей (лактозой). Патогенез болезни включает накопление галактозы и галактозо-1-фосфата в крови и разных тканях, выделение его с мочой, накопление в хрусталике производного галактозы галакти-тола. Затем происходит нарушение использования глюкозы в печени, почках, головном мозге вследствие угнетения активности фермента фосфоглюкомутазы. В крови обнаруживается снижение содержания глюкозы, а в моче –определённые аминокислоты (метионин, цистин и др).
Заболевание развивается после рождения при вскармливании младенца молоком, с которым поступает лактоза — источник неметаболируемой галактозы. Основными симптомами заболевания являются: желтуха новорожденных, рвота и понос, приводящие к обезвоживанию организма, постепенное развитие умственной отсталости, увеличение печени и селезенки, общая дистрофия, катаракта. При лабораторном обследовании обнаруживают галактозу и белок в моче, снижение активности галактозо-1-фосфатуридилтрансферазы в эритроцитах.
Без лечения больные погибают в первые месяцы жизни от сопутствующих инфекций или печеночной недостаточности, у выживших развиваются катаракта и умственная отсталость. Раннее лечение диетой (исключение из пищи лактозы) приводит к нормальному развитию детей.

- Мукополисахаридозы — это группа наследственных дефектов катаболизма гликозаминогликанов (ГАГ), преимущественно с аутосомно-рецессивным типом наследования. Популяционная частота их не уста-новлена. При мукополисахаридозах наблюдаются повышенная экскреция или внутриклеточное накоп-ление ГАГ вследствие нарушения их расщепления, обусловленного дефектами ферментов, осуществляю-щих этот процесс. Мутантные ферменты относятся к лизосомным гидролазам. Выделяют несколько ти-пов мукополисахаридозов. Рассмотрим мукополисахаридоз первого типа (синдром Гурлер), обусловлен-ный дефицитом фермента L-индуронидазы, ответственного за катаболизм кислых мукополисахаридов.
Дети с синдромом Гурлер рождаются без внешних изменений. В первые месяцы жизни черты лица ста-новятся грубыми, западает перено-сица, развиваются помутнение роговицы, тугоподвижность суставов, искривление позвоночника, увеличиваются печень и селезенка. Рот открыт. На втором году жизни выяв-ляются короткая шея, воронкообразная грудная клетка, паховые и пупочные грыжи, увеличение головы, языка и губ, мелкие, редко посаженные зубы, ограничение подвижности большинства суставов, прогрес-сирующая умственная отста-лость. Позже поражается сердце, развиваются глухота и слепота. Больные погибают обычно в возрасте до 10 лет.
Диагностика основана на изучении содержания кислых мукопо-лисахаридов в сыворотке крови.
Характеристика моногенных заболеваний, связанных с нарушением липидного обмена.
Наследственные дефекты обмена липидов подразделяют на две большие группы: сфинголипидозы и нарушения обмена липидов плазмы крови.
- Сфинголипидозы представляют собой болезни накопления сфинголипидов (одной из разновидностей гликолипидов), обусловленные дефектами ферментов, катализирующих их расщепление. Эти болезни встречаются редко (1:300000). Высокая частота болезни Тея-Сакса наблюдается только среди евреев-ашкенази (1:3600 новорожденных). Все сфинголипидозы имеют рецессивный тип наследования, аутосомный или сцепленный с Х-хромосомой.
Сфинголипиды являются важнейшими структурными компонентами мембран клеток, в частности, миелиновых оболочек нервных волокон, поэтому при нарушении их обмена поражается большинство жизненно важных органов, в том числе серое и белое вещество головного мозга.
Сфинголипидозы характеризуются выраженным клиническим полиморфизмом, касающимся и времени на-чала болезни и тяжести течения. Эти болезни проявляются прогрессирующими умственными и двигатель-ными расстройствами вследствие изменений головного мозга. Наблюдаются поражение костей, паренхи-матозных органов (печень, селезенка, почки), кожи и сетчатой оболочки глаза.

При болезни Тея-Сакса психомоторные нарушения начинают развиваться у детей с 4—6 месяцев. Они становятся апатичными, перестают интересоваться окружающим, наблюдается мышечная гипотония. К концу первого года развивается слепота, обусловленная атрофией зрительных нервов; интеллект снижается до уровня идиотии. Постепенно развиваются полная обездвижен-ность, судороги, не поддающиеся терапии. Смерть обычно наступает в 3—4 года.
Для диагностики применяют биохимические методы исследования гликолипидов.
- Гиперлипопротеинемии обусловлены нарушением обмена липидов плазмы крови вследст-вие дефектов ферментов или клеточных рецепторов. Липиды плазмы крови представляют собой большую группу соединений, в основном, жирных кислот, триглицеридов и холестерина. Повышенное содержание липидов в плазме крови может быть мультифакториальной природы или моногенно обус-ловленным дефектом с аутосомно-доминантным типом наследования. Частота гетерозигот моногенно обусловленных гиперлипо-протеинемий в популяции составляет 1:500. Ген картирован — 19р13.
Значение гиперлипопротеинемий и их моногенных форм определяется тем, что с метаболизмом этих соединений тесно связан патогенез атеросклероза и ишемической болезни сердца. Генетической особенностью гиперхолестеринемии с дефектом рецепторов является то, что повышенный уровень холестерина имеется и у гетерозигот (в 2-3 раза выше нормы) и гетерозиготы подвержены раннему (в 35—45 лет) развитию инфарктов миокарда. Моногенные гиперлипопротеинемий встречаются у небольшой части больных атеросклерозом.
Диагностика основана на определении липопротеинов сыворотки крови.
Наследственные дефекты обмена пуринов и пиримидинов.
Данную патологи. можно рассмотреть на примере синдрома Леша-Нихана.

- — обусловлен недостаточностью фермента гипоксантин-фосфорибозилтрансферазы (ГФРТ), который катализирует присоединение свободных пуриновых оснований (гуанина и гипоксантина) к нуклеотидам. Синдром встречается редко (1:300000 новорожденных), наследование идет по сцепленному с полом рецессивному типу.
При недостаточности фермента ГФРТ конечным продуктом превращения пуриновых оснований является мочевая кислота. Болезнь начинает развиваться в грудном возрасте, проявляясь мышечным гипертонусом, повышенной рефлекторной возбудимостью, олигофренией, склонностью ребенка к самоповреждениям. Высокое содержание мочевой кислоты и ее солей (диагностический признак), несмотря на усиленное выделение их с мочой, приводит к формированию камней в мочевыводящих путях, отложению солей мочевой кислоты в суставах.
Характеристика заболеваний, связанных с нарушением минерального обмена.
Примером такой патологии может служить расстройство обмена меди.
- Болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия) обусловлена генной мутацией, тормозящей синтез белка церулло-плазмина, обеспечивающего транспорт меди в организме. Тип наследования — аутосомно-рецессивный. Популяционная частота не установлена.
Соединения меди играют большую роль в обменных процессах. Ионы меди входят в состав многих ферментов митохондрий, участвующих в реакциях окисления. При недостатке церуллоплазмина повышается концентрация меди в крови и происходит отложение ее в тканях печени и мозга, вызывающее их дегенерацию. Заболевание чаще проявляется в школьном возрасте. Первыми симптомами могут быть увеличение печени и селезенки, нарушение функции печени, ЦНС, иногда почек, снижение количества эритроцитов, тромбоцитов и лейкоцитов в крови. Поражение печени сопровождается желтухой, рвотой, диспепсией, постепенно развивается цирроз. Поражения ЦНС сопровождаются снижением интеллекта, изменением поведения, дрожанием рук, нарушением глотания, повышением тонуса мышц. Диагностика основана на определении количества церуллоплазмина в сыворотке крови.
2.7. Наследственные заболевания, вызванные нарушением развития органов и тканей.
- Муковисцидоз (кистофиброз поджелудочной железы) обусловлен генной мутацией в 7-ой хромосоме (7q21-q31).Tиn наследования — аутосомно-рецессивный. Популяционная частота заболевания 1:2500 новорожденных. Это одно из самых распространенных наследственных заболеваний. Муковисцидоз представляет собой множественные поражения желез внешней секреции, проявляющиеся выделением секретов повышенной вязкости, что ведет к застойно обтурационным изменениям в соответствующих органах (легких, поджелудочной железе и кишечнике) с последующими воспалительными и склеротическими процессами. Легочные проявления болезни возникают на 1-2 году жизни и характеризуются рецидивирующими пневмониями и бронхитами с последующим развитием эмфиземы легких. Кишечные проявления муковисцидоза связаны с нарушением активности ферментов поджелудочной железы. Гнилостные процессы в кишечнике приводят к вздутию живота, появлению обильного жирного стула с резким гнилостным запахом. Иногда наблюдается картина кишечной непроходимости. В некоторых случаях развивается цирроз печени. Диагностика основана на клинической картине и изучении активности ферментов поджелудочной железы.
- Ахондроплазия (хондродистрофия) обусловлена генной мутацией, вызывающей отклонения в активности некоторых ферментов (5-нуклеотидазы, глюкозо-6-фосфатазы). Тип наследования аутосомно-доминантный. Популяционная частота 1:100000. 80% случаев болезни обусловлены но-выми мутациями. Ген картирован — 4р14-р16.
Ахондроплазия — одна из наследственных болезней костной системы. Она обусловлена аномальным ростом и развитием хрящевой ткани чаще всего в эпифизах трубчатых костей и основании черепа, результатом чего является резкое недоразвитие костей в длину. Характерными признаками заболевания являются низкий рост (120-130 см у взрослых) при сохранении нормальной длины туловища, большой череп с выступающим затылком, запавшая переносица. Конечности укорочены в основном за счет проксимальных отделов бедренной и плечевой костей, кисти широкие и короткие. Дети отстают в моторном развитии, интеллект, как правило, нормальный. Диагностика заболевания не представляет затруднений.
- Миодистрофия Дюшенна (МД) — тяжелое наследственное заболевание с повышенной активностью в плазме крови ряда мышечных ферментов, особенно креатинкиназы. Встречается с частотой 1:3500 новорожденных мальчиков. Наследование сцепленное с полом, рецессивное. Ген МД картирован в области Хр21 и детально изучен.
Для МД характерно раннее, в возрасте 3-5 лет, начало заболевания: нарастающая слабость в мышцах бедер и таза с постепенным переходом процесса на икроножные мышцы, мышцы верхнего плечевого пояса, спины, живота и др. Появляется утиная походка. Заболевание неуклонно прогрессирует, дети оказываются прикованными к постели с 10—11-летнего возраста. Наблюдается псевдогипертрофия икроножных и ягодичных мышц за счет замещения мышечной ткани соединительной и жировой. Часто развивается сгибательная мышечная контрактура бедренных и коленных суставов и суставов верхних конечностей вследствие атрофии мышц. Рано снижаются глубокие сухожильные рефлексы. Имеется тенденция к некоторому снижению умственных способностей. Продолжительность жизни больных 20-35 лет. Смерть обычно наступает от легочной инфекции или сердечной недостаточности из-за миокардиодистрофии. Диагностика основана на клинической картине и повышении активности креатинкиназы в сыворотке крови.
- Синдром фрагильной (ломкой) Х-хромосомы —тяжелое наследственное заболевание. Тип наследования — сцепленный с полом доминантный с неполной пенетрантностью (80%). Частота встречаемости 1:1500 новорожденных мальчиков. Мутантный ген картирован: он расположен в Xq27.
Заболевание характеризуется определенным типом умственной отсталости. Больные мужчины обладают характерными физическими признаками: большие яички, большие уши, выпуклый лоб и выступающие челюсти. При рождении некоторые имеют большую голову и повышенный вес. Речь характеризуется постоянными повторами, широко распространено заикание. Среди гетерозиготных женщин «нарушения психики» наблюдаются примерно в 30% случаев. В настоящее время этим синдромом объясняют до 20% всех случаев сильной или умеренной умственной отсталости. Для диагностики используют цитогенетический метод — в Х-хромосоме обнаруживают вторичное сужение. Описан терапевтический эффект фолиевой кислоты у таких больных.
2.8. Наследственные заболевания, вызываемые нарушениями свертывающей системы крови.
- Гемофилия А — тяжелое наследственное заболевание, обусловленное дефектом VIII фактором свертывания крови. Встречается с частотой 1:6500 мальчиков. Тип наследования — сцепленный с полом, рецессивный. Ген расположен в длинном плече Х-хромосомы (Xq28), порядок его нуклеотидов установлен.
Заболевание распознается обычно на 2—3-м году жизни, а в тяжелых случаях- при рождении (кровотечения из пупочного канатика, под- и внутрикожные кровоизлияния). Для заболевания характерен гематомный тип кровоточивости. Преобладают кровоизлияния в крупные суставы конечностей (коленные, локтевые, голеностопные), подкожные, внутри- и межмышечные гематомы, кровотечения при травмах и хирургических вмешательствах, наличие крови в моче. Поступление крови в полость суставов приводит к развитию стойкой тугоподвижности из-за остеоартрозов (развитие соединительной ткани в суставах).
- Гемофилия В — тяжелое наследственное заболевание, обусловленное снижением активности IX фактора свертывания крови. Популяционная частота не установлена. Тип наследования — сцепленный с полом, рецессивный. Ген картирован Xq27. Клинические проявления заболевания сходны с таковыми при гемофилии А. Диагностика основывается на исследовании соответствующих факторов свертывания крови. При лечении вводят недостающие факторы свертывания крови.
- Гемоглобинопатии — заболевания, связанные с нарушением структуры молекулы гемоглобина. Нормальный гемоглобин человека (НЬА) состоит из двух а-цепей и двух b-цепей. Большую часть структурных вариантов НЬ составляют одиночные замены аминокислот, в основе которых лежит замена одного азотистого основания другим с изменением кода триплета.
- Наиболее известной формой аномальных гемоглобинов является серповидно-клеточная анемия, при которой в 6-м положении а-цепи глутаминовая кислота замещена валином (HbS). Эта замена обусловливает пониженную растворимость гемоглобина, и у гомозигот эритроциты приобретают серповидную форму. Гетерозиготные носители HbS в обычных условиях клинически здоровы.
У гомозигот с раннего возраста развивается характерная картина хронической гипоксии и анемий, обусловленная слабой способностью HbS переносить кислород и преждевременным гемолизом и распадом эритроцитов. HbS часто обнаруживается у населения регионов с широким распространением тропической малярии, так как даже гетерозиготы по HbS невосприимчивы к малярии.
- Талассемии обусловлены мутациями глобиновых генов, приводящих к уменьшенному содержанию его или полному отсутствию. Причиной а-талассемий служат полные делеции НЬ а-генов. Таких генов четыре, и от количества делеции генов зависит тяжесть заболевания. Они расположены в 16-ой хромосоме — 16р13.3. При р-талассемиях имеется дефицит синтеза р-глобина. Генетические дефекты разнообразны (делеции и нарушение работы функциональных генов). При выраженных талассемиях наблюдается гемолитическая анемия. У гетерозиготных носителей гена р-талассемий выраженные признаки анемии обычно не выявляются.
Помимо биохимических методов диагностики генных болезней все большее распространение находят методы рекомбинантной ДНК.
Хромосомные болезни.
Хромосомные болезни — это группа заболеваний, вызываемых изменениями числа (геномные мутации) или структуры (хромосомные аберрации) хромосом, видимыми в световой микроскоп.
Существует очень много разнообразных аномалий кариотипа. Хромосомные аберрации и изменения количества хромосом могут возникать на разных этапах. Если они имеются уже в гаметах родителей, то аналогичная аномалия будет наблюдаться во всех клетках развивающегося организма (полный мутант).
Хромосомные аномалии могут возникать и в процессе эмбрионального развития при дроблении зиготы. В норме каждый бластомер содержит одинаковый набор хромосом, идентичный набору зиготы. Однако в некоторых случаях возможно нарушение расхождения хромосом, вследствие чего в разные бластомеры попадет неодинаковое их количество (один бластомер с моносомией, а другой — с трисомией). При последующем дроблении возникают две клеточные линии (клоны), сохраняющие особенности аномального кариотипа. В зависимости от стадии, на которой произошло нарушение, и интенсивности размножения клеток число этих клеточных популяций может быть различным. Остальные клетки, ведущие начало от нормальных бластомеров, будут иметь неизмененный кариотип. Такое явление называется генетическим мозаицизмом. Мозаичные организмы могут содержать несколько (2, 3, 4 и более) клеточных клонов с различными кариотипами. Это явление может сопровождаться патологией всего организма, либо отдельных его органов и систем. При незначительном количестве аномальных клеток фенотипические проявления могут не обнаруживаться.
В основе хромосомных болезней лежат синдромы, связанные с нарушением плоидности, изменениями числа хромосом или нарушением их структуры. Нарушение плоидности у детей представлено лишь синдромом триплоидии (дети погибают в первые часы или дни после рождения). Синдромы трисомий — наиболее частая форма хромосомной патологии у человека. Полная моносомия, совместимая с жизнью, наблюдается только по Х-хромосоме. В основе синдромов, обусловленных структурными нарушениями хромосом, лежат либо частичные трисомий, либо частичные моносомии, либо их сочетания. Хромосомные болезни встречаются довольно часто. Частота хромосомных болезней у живорожденных детей составляет примерно 2, 4 случая на 1000 родившихся. Большинство хромосомных аномалий (полиплоидии, гаплоидии, трисомий и моносомии по первым парам крупных хромосом являются несовместимыми с жизнью. Такие эмбрионы или плоды элиминируются из организма матери на ранних или более поздних сроках беременности.
Клинически почти все хромосомные болезни проявляются:
а) нарушением интеллектуального развития;
б) множественными врожденными пороками.
Эти нарушения являются тяжелыми и касаются физического, психического и полового развития. В нас-тоящее время внутриутробная диагностика хромосомных заболеваний возможна при исследовании околоплодных вод, что позволяет предупредить рождение детей с хромосомными заболеваниями. Причинами, приводящими к хромосомным мутациям могут быть физические, биологические и химические факторы. Мутации в яйцеклетках женщины могут возникать во внутриутробном развитии, но риск развития хромосомных заболеваний больше среди пожилых женщин (риск рождения ребёнка с трисомией для женщин старше 45 лет в 60 раз превышает эту патологию у женщин в возрасте 19-24 лет). Очень молодые женщины также имеют повышенный риск рождения ребёнка с хромосомными заболеваниями.
Окончательный диагноз хромосомных болезней устанавливается цитогенетическими методами.
3.1. Аутосомные трисомии.
Наиболее часто у человека встречаются трисомии по 13-й, 18-й и 21-й паре хромосом.
с массой тела значительно ниже нормы (2500 г).
у них наблюдается умеренная микроцефа-лия,
недоразвитие различных отделов ЦНС,
низкий скошенный лоб;
суженные глазные щели, расстояние между которыми уменьшено;
микрофтальмия,
помутнение роговицы,
запавшее переносье,
широкое основание носа,
широко расположенные и деформированные ушные раковины с гипоплазированным козелком.
у половины больных определяется попе-речная ладонная борозда.
Одним из наиболее типичных признаков этого синдрома является двухсторонняя расщелина верхней губы и неба.
Отмечаются аномалии опорно-двигатель-ного аппарата: полидактилия, синдактилии, флексорное положение кистей) и короткая шея.
У 80% новорожденных встречаются пороки развития сердца:
дефекты межжелудочковой и межпредсердной перегородок,
транспозиции сосудов и др.
Наблюдаются пороки развития поджелудочной железы: она увеличена, хвост утолщен, иногда раздвоен, сращен с селезенкой, имеются фиброкистозные изменения.
Почки часто увеличены,
наблюдается повышенная дольчатость и кисты в корковом слое.
Дети с синдромом Патау живут недолго. По данным литературы, 95% таких больных умирают до года, более трех лет живут лишь единицы. Все выжившие дети с синдромом Патау — глубокие идиоты.
- Синдром Эдвардса (синдром трисомии 18) встречается с частотой примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей. Для женщин старше 45-ти лет риск родить больного ребенка составляет 0,7%.
Цитогенетически синдром Эдвардса представлен простой трисомией 18, значительно реже встречаются мозаичные формы и как исключение — транслокационные.
Аномалии скелета:
грудина короткая,
межреберные промежутки уменьшены,
грудная клетка широкая и короткая.
В 80% случаев наблюдается аномальное развитие стопы: пятка резко выступает,
свод провисает (стопа-качалка).
Изредка наблюдаются спинно-мозговые грыжи и односторонние расщелины губы.
Из пороков внутренних органов наиболее часто наблюдаются:
пороки сердца и крупных сосудов: нарушения развития межжелудочковой перегородки, аплазии одной створки клапанов аорты и легочной артерии.
В головном мозге отмечаются гипоплазия мозжечка и мозолистого тела, изменения структуры олив.
В пищеварительной системе могут наблюдаться дивертикул Меккеля, атрезии пищевода. У 10% больных встречается подковообразная почка.
При дерматоглифическом анализе обнаруживают: поперечную четырехпальцевую борозду, увеличение числа дуг на пальцах, слабо выраженный узор на мизинцах.
Продолжительность жизни детей с синдромом Эдвардса невелика: 60% детей умирают до 3 месяцев; до года доживает лишь 1 ребенок из 10.
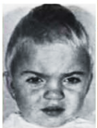
Наиболее распространенными цитогенетическими формами синдрома Дауна являются простая трисомия, транслокационная форма и мозаицизм. За возникновение фенотипических проявлений синдрома Дауна отвечает лишь небольшой участок длинного плеча 21-й хромосомы (21q+), и каким бы образом он не был удвоен, развивается типичная клиническая картина.
Дети с синдромом Дауна рождаются с несколько сниженным весом (3167 г). Для больных характерна: округлой формы голова с уплощенным затылком,
лоб скошен, узкий, лицо плоское.
типичен эпикант, плоская спинка носа,
косой разрез глазных щелей,
пятна Брушфильда (светлые пятна на радужке),
толстые губы, утолщенный язык с глубокими бороздами, выступающий изо рта,
маленькие недоразвитые низко расположенные ушные раковины,
недоразвитая верхняя челюсть,
высокое небо,
неправильный рост зубов,
короткая шея.
Из пороков внутренних органов наиболее типичны:
пороки сердечно-сосудистой системы (дефекты межжелудочковой или межпредсердной перегородок и др.);
органов пищеварения (атрезии и стенозы различных отделов).
У детей младшего возраста резко выражена мышечная гипотония, а у детей старшего возраста часто обнаруживается катаракта. Для детей с синдромом Дауна характерна умственная отсталость: в основном это имбецильность (65-90%), дебильность и идиотия диагностируются примерно в равном соотношении. Дерматоглифические особенности при болезни Дауна представлены четерехпальцевой поперечной бороздой (у 45% больных) и главным ладонным углом свыше 57°.
Продолжительность жизни при синдроме Дауна значительно ниже (36 лет), чем в популяции.
3.2. Характеристика синдромов, связанных с частичными трисомиями.
Помимо полных трисомий, описано довольно много синдромов, связанных с частичными трисомиями.
- Синдром трисомий по короткому плечу хромосомы 9. Синдром трисомий 9р+ — наиболее частая форма частичных трисомий. Описано не менее 150 больных с такой патологией.
Для больных с трисомией 9р+ характерны умственная отсталость, задержка роста, микроцефалия, антимонголоидный разрез глазных щелей, глубоко посаженные глаза, опущенные углы рта, нос с характерным округлым кончиком, низко расположенные оттопыренные ушные раковины, недоразвитие дистальных фаланг пальцев рук и ногтей. Часто наблюдаются выступающие лобные кости, повышенная обволошенность, пятна цвета «кофе с молоком» на коже, эпикант, косоглазие, высокое дугообразное небо, короткая шея, сколиоз, сращение пальцев на стопах. Примерно в четверти случаев обнаруживаются врожденные пороки сердца, иногда почек.
Прогноз для жизни сравнительно благоприятный: описаны больные, достигшие преклонного возраста.
3.3. Синдромы частичных моносомий. Описано большое количество синдромов, причинами которых являются частичные моносомии.
- Синдром Вольфа-Хиршхорна (4р-) обусловлен делецией короткого плеча хромосомы 4 Популяционная частота заболевания около 1:100000. Дети с синдромом Вольфа-Хиршхорна обычно рождаются от молодых родителей, доношенными, но со значительным снижением веса (2000 г). Для таких детей характерна резкая задержка физического и психомоторного развития. У них умеренно выраженная: микроцефалия,
клювовидный нос,
выступающее надпереносье,
деформированные низко расположенные ушные раковины,
впереди ушной раковины часто имеются вертикальные складки кожи,
гипотония мышц,
значительное снижение реакции на внешние раздражения,
судорожные припадки.
Часто отмечаются расщелины верхней губы и неба, деформации стоп, аномалии глазных яблок, эпикант и маленький рот с опущенными углами.
Из внутренних органов чаще всего поражается сердце (разнообразные пороки развития), примерно в половине случаев — почки (гипоплазия, кисты), иногда — желудочно-кишечный тракт (гипоплазия желчного пузыря, подвижная слепая кишка).
- Синдром «кошачьего крика» (5р-) обусловлен делецией короткого плеча 5-й хромосомы. Дети с этим синдромом рождаются у родителей обычного возраста. Популяционная частота синдрома примерно 1:45000.
Наиболее характерными для синдрома 5р- являются:
специфический плач («кошачий крик»),
умственное и физическое недоразвитие,
микроцефалия,
низко расположенные,
иногда деформированные ушные раковины,
лунообразной формы лицо,
эпикант,
антимонголоидный разрез глазных щелей,
косоглазие и гипотония мышц.
Иногда наблюдаются аномалии глаз (атрофия зрительного нерва, очаги депигментации сетчатки). Наиболее постоянный признак синдрома — «кошачий крик» обусловлен изменениями гортани: сужением, мягкостью хрящей, отечностью или необычной складчатостью слизистой, уменьшением надгортанника. Изменения других органов и систем неспецифичны.
Продолжительность жизни пациентов значительно снижена, только около 14% больных переживают возраст 10 лет.
- Синдром Орбели (13 q-) обусловлен делецией длинного плеча 13-й хромосомы сегментов 13q22-q31. Популяционная частота синдрома не установлена,
Дети с синдромом Орбели рождаются с низким весом (2200 г). Клинически этот синдром сопровождается аномалиями развития всех систем органов. Для этого синдрома характерна:
микроцефалия,
лоб переходит в нос, не образуя носовой вырезки,
эпикант,
антимонголоидный разрез глаз,
широкая спинка носа,
высокое небо,
низко расположенные деформированные ушные раковины.
Весьма характерны признаки поражения глаз (микрофтальмия, иногда анофтальмия, косоглазие, катаракта, ретинобластома), опорно-двигательного аппарата (короткая шея, гипо- или аплазия первого пальца кисти и пяточной кости, синдактилии кистей и стоп), атрезии прямой кишки и ануса. Часты пороки развития сердца, почек, головного мозга. Для всех детей с синдромом Орбели характерна глубокая олигофрения, возможны потеря сознания, судороги.
Большинство больных синдромом 13q- погибают на первом году жизни.
3.4. Характеристика синдромов, связанных с изменениями числа половых хромосом.
- Трисомия по Х-хромосоме.
Впервые цитогенетика синдрома трисомии была описана в 1959 г. А.Джекобсом. Его частота составляет 1:1000 —1:2 000 новорожденных девочек.
Как правило, физическое и психическое развитие у женщин с этим синдромом не имеет отклонений от нормы. Это объясняется тем, что у них инактивируются две Х-хромосомы, а одна продолжает функционировать, как у нормальных женщин. Изменения в кариотипе, как правило, обнаруживаются случайно при обследовании.
Умственное развитие также обычно нормальное, иногда на нижних границах нормы. Лишь у некоторых женщин отмечаются нарушения со стороны репродуктивной функции (различные нарушения цикла, вторичная аменорея, ранняя менопауза).
При тетрасомиях X встречается:
- высокий рост,
- телосложение по мужскому типу,
- эпикант,
- гипертелоризм,
- уплощенное переносье,
- высокое небо,
- аномальный рост зубов,
- деформированные и аномально расположенные ушные раковины,
- клинодактилия мизинцев,
- поперечная ладонная складка.
У этих женщин описаны различные нарушения менструального цикла, бесплодие, преждевременный климакс.
Снижение интеллекта от пограничной умственной отсталости, различ-ных степеней олигофрении описано у двух третей больных. Среди женщин с полисемией X увеличена частота психических заболеваний (шизофрения, маниакально-депрес-сивный психоз, эпилепсия).
При пентасомиях X увеличивается число патологических сим-гтомов у больных, нарастает степень умственной отсталости.
- Синдром Клайнфельтера.
Впервые этот синдром был описан в 1942 г. Г. Ф. Клайнфельтером, а в 1959 г. П. Джекобе и Дж. Стронг подтвердили хромосомную этиологию данного заболевания (47, ХХУ).
Синдром Клайнфельтера наблюдается у 1 из 500—700 новорожденных мальчиков; у 1—2,5% мужчин, страдающих олигофренией (чаще при неглубоком интеллектуальном снижении); у 10 % мужчин, страдающих бесплодием.
В период новорожденности заподозрить этот диагноз невозможно. Основные клинические проявления манифестируют в пубертатом периоде. Классическими проявлениями этого заболевания считаются:
- высокий рост,
- евнухоидное телосложение,
- гинекомастия, но все эти симптомы одновременно встречаются лишь в половине случаев.
В среднем рост больных с этим синдромом не отличается от здоровых мужчин, гинекомастия обнаруживается у 25 % пациентов. Наружные половые органы сформированы по мужскому типу, размеры полового члена нормальные, яички резко уменьшены в размерах (их диаметр не более 1,5 см). Именно микрогонадизм является главным клиническим признаком этого синдрома. Объем эякулята снижен, наблюдается постоянная азооспермия. Мужчины, страдающие синдромом Клайнфельтера,бесплодны.
- скудность или отсутствие оволосения а лице, в подмышечных впадинах, на груди.
- На лобке оволосение располагается по женскому типу.
- часто отмечают уплощенный затылок,
- гипертелоризм,
- эпикант,
- выступающие надбровные дуги,
- высокое небо,
- аномальный рост зубов,
- клинодактилию мизинцев.
- Умственная отсталость отмечается в 25—50 % случаев. Степень умственной отсталости колеблется от пограничных состояний до дебильности различной тяжести.
- Больным свойственны неустойчивость внимания,
- повышенная утомляемость и отвлекаемость,
- снижение работоспособности,
- повышенная внушаемость,
- снижение инициативной способности к длительному волевому действию,
Дальнейшее увеличение числа Х-хромосом (48, XXXV, 49, ХХХУ) в кариоти-пе ведет к более выраженному интеллектуальному эффекту и к наличию более широкого спектра симптомов пациентов.
- Синдром дисомии по Y-хромосоме.
Этот синдром впервые в 1961 г. описали А. А. Сандберг с соавторами, определив его цитогенетику — 47, ХУУ.
Частота этого синдрома среди новорожденных мальчиков составляет 1:840 и возрастает до 10 % у высокорослых мужчин (выше 200 см).
У большинства больных отмечается ускорение роста в детском возрасте. Средний рост у взрослых мужчин составляет 186 см. Они в большинстве случаев не отличаются от нормальных индивидов по физическому и умственному развитию. Заметных отклонений в половой и в эндокринной сферах и в плодовитости нет. Хотя не исключены некоторые особенности поведения у таких лиц — они склонны к агрессивным и криминальным поступкам. Однако 30— 40 % пациентов имеют определенные симптомы — грубые черты лица, выступающие надбровные дуги и переносье, увеличенная нижняя челюсть, высокое небо, аномальный рост зубов с дефектами зубной эмали, большие ушные раковины, патология коленных и локтевых суставов (валыусная девиация). Иногда отмечаются различные нарушения половой сферы (крипторхизм, дисплазия гениталий, гипогонадизм, бесплодие), пограничная умственная отсталость или легкая дебильность.
Продолжительность жизни у таких больных не отличается от популяционной.
- Синдром Шерешевского-Тернера.
Впервые этот синдром был описан в 1925 г. русским ученым профессором Н. А. Шерешевским. В 1938 г. также клинически, но более полно этот синдром был описан Ц. Тернером. Этиология заболевания (моносомия по Х-хромосоме) была раскрыта Ч. Форцом в 1959 г.
Частота синдрома Шерешевского—Тернера среди новорожденных девочек составляет 1:2 000 — 1:5000.
Наиболее часто при цитогенетическом исследовании обнаруживается:
- кариотип 45, ХО (в 45 % случаев),
- встречаются делеции короткого или длинного плеча Х-хромосомы,
- изохромосомы,
- кольцевые хромосомы,
- различные варианты мозахизма (30—40 %).
Рождение девочек с этим синдромом не связано с возрастом родителей. Беременность протекает, как правило, без осложнений, нормальной продолжительности, но дети рождаются с умеренной пренатальной гипоплазией.
В периоде новорожденности у больных с кариотипом 45, ХО наиболее убедительными признаками этого заболевания является:
- короткая с кожными складками шея (шейный птеригиум),
- лимфатический отек кистей и стоп (50% случаев).
Обращает на себя внимание наличие множественных малых аномалий развития:
В пубертатном периоде наблюдается:
- отсутствие формирования вторичных половых признаков (почти нет железистой ткани молочных желез, соски недоразвиты, ареолы узкие, отсутствует оволосение на лобке и в подмышечных впадинах),
- первичная аменорея.
- Гонады больных представлены соединительно-тканными тяжами. В них находят либо совершенно недифференцированные рудименты, половая принадлежность которых не устанавливается, либо рудименты женских гонад без овариальных элементов.
- редко обнаруживаются примордиальные фолликулы. Деторождение таких больных невозмож-но.
Из внутренних органов наиболее часто поражается сердце (в 25 % случаев):
чаще это коарктация аорты, реже — персистирование артериального протока и другие пороки. Другим частым поражаемым органом являются почки. Наиболее частый порок — подковообразная почка, реже — гипоплазия, пиелоэктазия, гидронефроз.
Интеллект больных с синдромом Шерешевского—Тернера сохранен, однако для них характерен своеобразный житейский практицизм, подчиняемость, узость интересов, малая продуктивность мышления.
При мозаичных формах отмечается стертая клиническая картина. У части больных нормально развиты вторичные половы: признаки, имеют место менструации. У некоторых больных возможно деторождение.
4. Принципы терапии наследственной патологии человека. Можно выделить три подхода к лечению наследственных болезней и болезней с наследственной предрасположенностью: симптоматическое, патогенетическое, этиологическое.
5.1. Симптоматическое лечение применяют при всех наследственных болезнях:
при болях — аналгетики,
при психических заболеваниях — успокаивающие,
при муковисцидозе — вещества, разжижающие слизь,
при воспалительных процессах — антибиотики и т.п.
При врожденных пороках развития широко применяется хирургическое симптоматическое лечение (удаление шестого пальца, полипов, катаракты, устранение пороков развития сердца и др.).
5.2. Патогенетическое лечение в настоящее время интенсивно разрабатывается для болезней обмена веществ, обусловленных генными мутациями. Оно может проводиться по следующим направлениям.
Коррекция обмена: ограничение или исключение неметаболируемого вещества из пищи (фенилаланина) и освобождение от продуктов обмена путем усиленного их выведения, плазмофереза или гемосорбции (фенилпировиноградная кислота). Метаболическая ингибиция применяется в тех случаях, когда надо снизить интенсивность синтеза накапливаемого при наследственной болезни субстрата (например, мочевой кислоты при подагре). Заместительная терапия применяется при наличии у больного аномальных ферментов, не обеспечивающих выработку продукта, например, введение тироксина при гипотиреозе, гормона роста при карликовости, инсулина — при диабете, некоторых коферментов и ферментов — при ферментопатиях, угнетение синтеза ферментов при их излишках и др.
5.3. Этиологическое лечение является наиболее оптимальным, поскольку устраняет причину заболевания и радикально его излечивает. Для этиологического лечения применяются методы генной инженерии, которые в настоящее время выходят за рамки экспериментов.

0 Комментариев